
Guillain-Barré-Syndrom
Polyradikuloneuritis
Das Guillain-Barré-Syndrom (GBS) ist eine akut bis subakut, d. h. über Tage bis maximal vier Wochen auftretende Entzündung von meist mehreren Nerven des peripheren Nervensystems und der Nervenwurzeln („Polyradikuloneuritis“). Es ist mit einer Erkrankungsrate von 1 – 2 pro 100.000 pro Jahr zur Zeit bei uns die häufigste Ursache einer akuten schlaffen Tetraparese, also einer Lähmung der Muskulatur von Beinen und Armen. Auch Nerven im Kopfbereich oder am Rumpf können betroffen sein. Das zentrale Nervensystem (Gehirn, Rückenmark) hingegen ist meist nicht beteiligt.
Zu Beginn kommt es zu sensiblen Missempfindungen und Taubheitsgefühlen an Fingern und Zehen. Dumpf ziehende Schmerzen in der Lendenwirbelsäule und den Flanken können sich dazu gesellen.
Das Vollbild der Erkrankung wird durch relativ symmetrische, innerhalb weniger Tage bis zu 4 Wochen von distal nach proximal aufsteigende (von den Zehen und Fingern in Richtung Rumpf), schlaffe Paresen mit Reflexverlust bestimmt, die in etwa zeitgleich Arme und Beine betreffen. Eine beidseitige Lähmung der Gesichtsmuskulatur („Fazialisparese“) ist oft frühzeitig nachweisbar, ebenso wie eine Schwäche der Kau- und Schluckmuskulatur. Aufgrund des Mitbefalls der Atemmuskulatur müssen etwa 25 – 30 % aller Patienten künstlich beatmet werden. Bei etwa zwei Drittel der Patienten sind die Fasern des autonomen Nervensystems betroffen, wobei seltene, aber potenziell lebensbedrohliche Herzrhythmusstörungen besonders beachtet werden müssen, da sie zumindest für einen Teil der Sterblichkeit von 3 – 5 % verantwortlich gemacht werden können.
Klassifikation
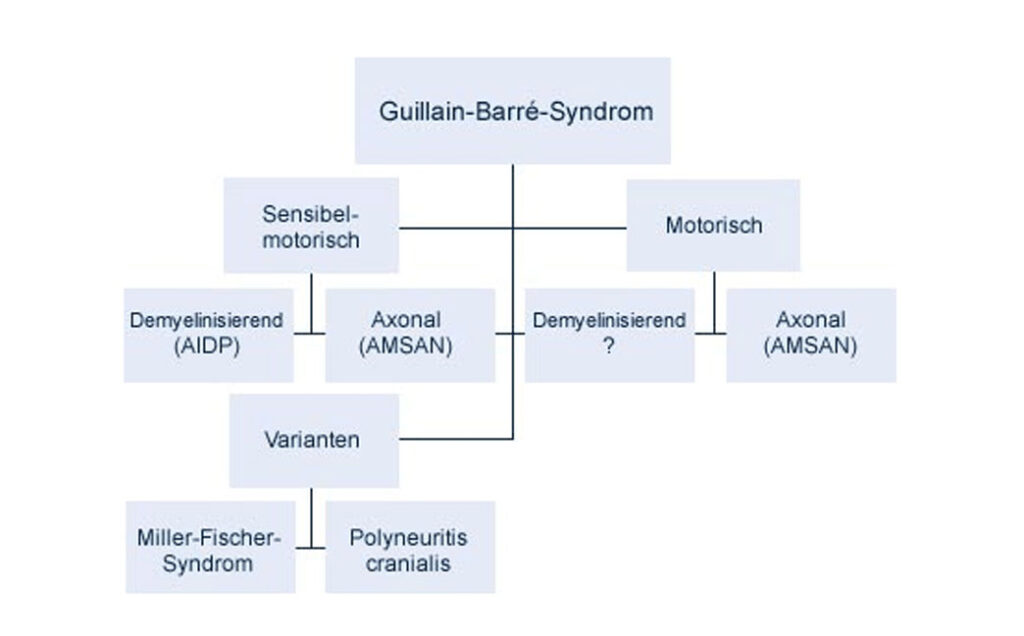
Unter dem Begriff „GBS“ werden verschiedenartige Erkrankungen zusammengefasst. Das „klassische“ GBS, die akute entzündliche demyelinisierende Polyradikuloneuropathie (AIDP), stellt mit 60 – 90 % die häufigste Form dar.
In 5 – 10 % liegt dem GBS eine axonale Neuropathie zugrunde (akute motorisch-sensible axonale Neuropathie, AMSAN). Eine saisonal in Epidemien auftretende Variante (akute motorische axonale Neuropathie, AMAN) wurde überwiegend bei Kindern und jungen Erwachsenen in China beschrieben, scheint aber auch in Indien und Südamerika vorzukommen. Etwa 2 – 3 % leiden an der Variante Miller-Fisher-Syndrom (MFS), das durch Augenmuskellähmung („Ophthalmoplegie“), Ataxie und Verlust der Muskeleigenreflexe („Areflexie“) gekennzeichnet ist und dessen Übergang zur AIDP fließend sein kann.
Ursache
Die Ursache des GBS wird als Folge fehlgeleiteter Immunreaktionen angesehen, die gegen Bestandteile der Markscheiden („peripheres Myelin“) oder der Nervenzellfortsätze („Axone“) gerichtet sind.
Etwa 70 % aller Patienten haben vor Ausbruch der Erkrankung einen grippalen oder gastrointestinalen Infekt durchgemacht, wobei der gramnegative Durchfallerreger Campylobacter jejuni am häufigsten mit dem GBS assoziiert ist. Es gibt auch GBS-Fälle in Folge einer SARS-CoV-2-Infektion. Wahrscheinlich wird die Bildung pathogen wirksamer Autoantikörper durch die Infektion getriggert und ist durch eine Kreuzreaktion mit Bestandteilen von peripheren Nerven bedingt („molecular mimicry“). Solche zirkulierenden Antikörper gegen bestimmte Nervenbestandteile lassen sich bei bis zu 30 % aller Patienten nachweisen, die Bestimmung dieser Antikörper ist aber abgesehen von den GQ1b-Antikörper beim Miller-Fisher-Syndrom für die Routinediagnostik nicht geeignet.
Diagnose
Die Diagnose des GBS wird klinisch gestellt und durch die Befunde der Liquoruntersuchung und der neurophysiologischen Diagnostik unterstützt.
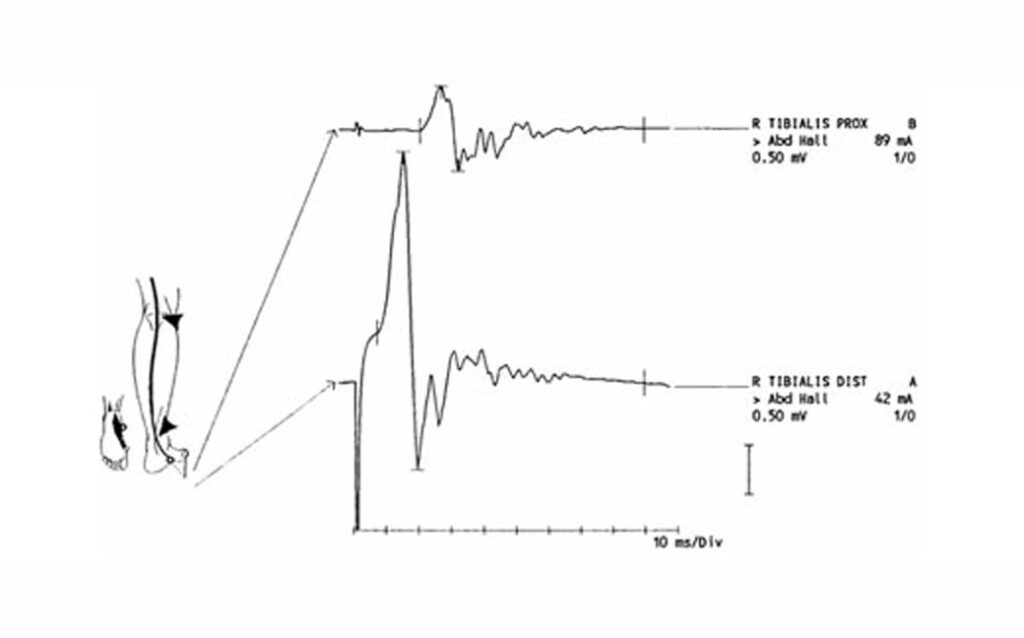
Diagnostische Kriterien sind seit 1978 verfügbar und basieren im wesentlichen auf einer über maximal 4 Wochen progrediente schlaffe Lähmung aller vier Extremitäten und fehlenden oder abgeschwächten Muskeleigenreflexen (Tabelle 1). Der typische Liquorbefund besteht in der „albuminozytologischen Dissoziation“ mit einer fehlenden oder nur mäßigen Zellzahlerhöhung (meistens < 10/ l, selten < 50/ l) und einer deutlichen Erhöhung des Liquoreiweisses, ist allerdings bei der Hälfte der Patienten erst eine Woche nach Krankheitsbeginn nachweisbar. Große Bedeutung kommt der neurophysiologischen Diagnostik zu, mit deren Hilfe zwischen demyelinisierenden und primär axonalen Formen unterschieden werden kann und für die ebenfalls Kriterien verfügbar sind. In den meisten Fällen finden sich die für eine demyelinisierende- Neuropathie typischen Veränderungen wie verlängerte F-Wellen-Latenz, verlängerte distal-motorische Latenz, verzögerte Nervenleitgeschwindigkeit, temporale Dispersion und Leitungsblockierungen.
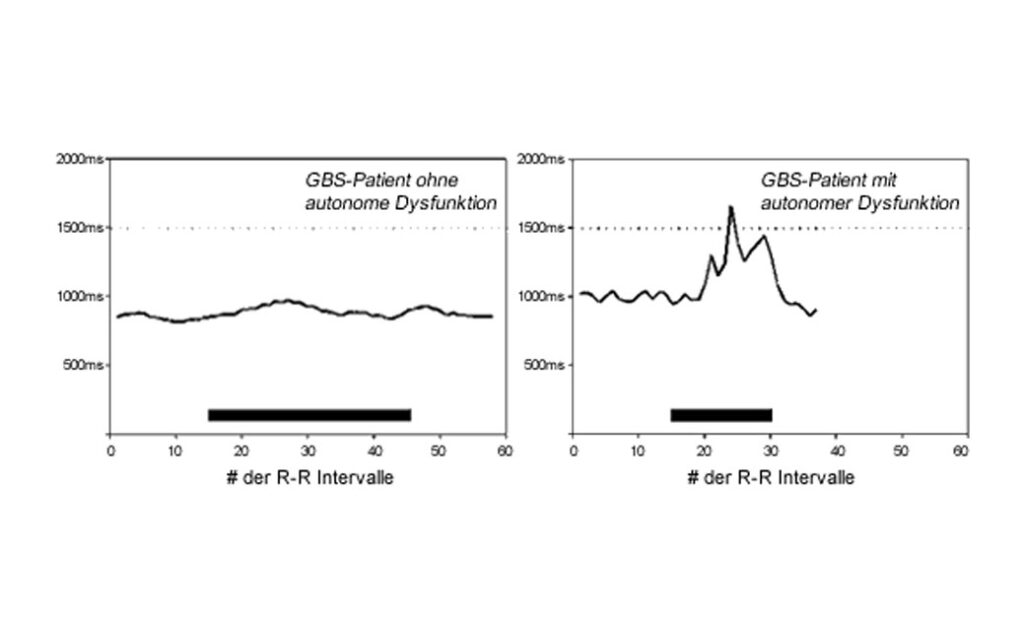
Zur Diagnostik potenziell bedrohlicher Bradyarrhythmien und zur Risikoabschätzung hat sich der Bulbusdruckversuch bewährt.
Die Diagnose des GBS wird klinisch gestellt und durch die Befunde der Liquoruntersuchung und der neurophysiologischen Diagnostik unterstützt. Diagnostische Kriterien sind seit 1978 verfügbar und basieren im wesentlichen auf einer über maximal 4 Wochen progrediente schlaffe Lähmung aller vier Extremitäten und fehlenden oder abgeschwächten Muskeleigenreflexen (Tabelle 1). Der typische Liquorbefund besteht in der „albuminozytologischen Dissoziation“ mit einer fehlenden oder nur mäßigen Zellzahlerhöhung (meistens < 10/ l, selten < 50/ l) und einer deutlichen Erhöhung des Liquoreiweisses, ist allerdings bei der Hälfte der Patienten erst eine Woche nach Krankheitsbeginn nachweisbar. Große Bedeutung kommt der neurophysiologischen Diagnostik zu, mit deren Hilfe zwischen demyelinisierenden und primär axonalen Formen unterschieden werden kann und für die ebenfalls Kriterien verfügbar sind. In den meisten Fällen finden sich die für eine demyelinisierende- Neuropathie typischen Veränderungen wie verlängerte F-Wellen-Latenz, verlängerte distal-motorische Latenz, verzögerte Nervenleitgeschwindigkeit, temporale Dispersion und Leitungsblockierungen.
Diagnostische Kriterien des Guillain-Barré-Syndroms
- Obligate Kriterien
- progrediente Muskelschwächemehr als einer Extremität mit maximaler Ausprägung innerhalb von vier Wochen
- abgeschwächte oder fehlende Muskeleigenreflexe
- Ausschluss anderer Ursachen (siehe Tabelle 2)
- Unterstützende Kriterien
- Klinische Befunde
- relativ symmetrische Ausprägung der Paresen
- gering ausgeprägte sensible Symptome
- Hirnnervenbeteiligung (besonders beidseitige Fazialisparese)
- autonome Funktionsstörungen (Tachy-/Bradykardie, Asystolie, Hypo-/Hypertonie)
- Fehlen von Fieber zu Beginn der neuropathischen Symptome
- Liquorbefunde
- erhöhtes Liquoreiweiß in der zweiten Krankheitswoche
- Zellzahl < 10/ l (selten < 50/ l)
- Elektrophysiologische Befunde (für AIDP)
- verzögerte Nervenleitgeschwindigkeit bzw. Leitungsblock
- verzögerte distal-motorische Latenz
- verzögerte F-Wellen-Latenz
- Befunde, die an der Diagnose zweifeln lassen
- Blasen- und Mastdarmstörung bereits zu Krankheits
- beginnsensibles Querschnittsniveau
- fortbestehende Asymmetrie der Muskelschwäche
- Klinische Befunde
Differentialdiagnose
Differentialdiagnose des Guillain-Barré-Syndroms
- Neuroborreliose
- Akut beginnende chronische Polyneuritis (CIDP)
- Akute Pandysautonomie
- Andere Polyneuropathien (vaskulitisch, paraneoplastisch, Porphyrie, „critical-illness“)
- Biologische Toxine (Botulismus, Diphterie)
- Intoxikationen mit Schwermetallen, Hexacarbone, Organophosphate
- Akute Motoneuron-Erkrankungen (Poliomyelitis, Rabies)
- Hirnstamminfarkt mit Locked-in-Syndrom
- Enzephalomyelitis mit Hirnstammbeteiligung
- Rückenmarkskompression
- Querschnittsmyelitis
- Akute nekrotische Myelopathie
- Hypo-/Hyperkaliämie
Verlauf und Prognose
Definitionsgemäß wird das Krankheitsmaximum innerhalb von vier Wochen erreicht, wobei 50 % der Patienten innerhalb von einer und 80 % der Patienten innerhalb von zwei Wochen die maximale Ausprägung der Lähmungserscheinungen erleben.
Nach einer Plateauphase von zwei bis vier Wochen kommt es zur Rückbildung der Symptome, die in der Regel einige Monate in Anspruch nimmt und bei etwa 75 % zu einer weitgehenden funktionellen Restitution führt. Neurologische Langzeitdefizite lassen sich bei immerhin 60 % aller Patienten feststellen, etwa 5 % bleiben schwer behindert. Faktoren, die mit einer ungünstigen Prognose assoziiert sind, sind höheres Lebensalter (> 50 Jahre), rascher Beginn (Vollbild innerhalb von 7 Tagen), Notwendigkeit der maschinellen Beatmung, reduzierte distale Amplitude des Muskelsummenaktionspotenzials bzw. fehlende Reizantworten bei der motorischen Neurographie und vorangegangene Durchfallerkrankung, insbesondere eine Infektion mit Campylobacter jejuni. Die Mortalität beträgt selbst in spezialisierten Zentren etwa 3 – 5 %. Bei weniger als 2 % aller Fälle kann es nach Monaten oder Jahren zu einer neuerlichen Krankheitsepisode kommen. Bei mehreren Rezidiven oder einer Progressionsphase, die länger als 4 Wochen dauert, muss eine chronisch-rezidivierende Verlaufsform, die chronisch-entzündliche demyelinisierende Polyradikuloneuropathie (CIDP, chronische Polyneuritis) vermutet werden.
Behandlungskonzept
Immun- und Plasmapherese-Therapie
Mit den Fortschritten der intensivmedizinischen Therapie haben sich die Behandlungsmöglichkeiten des GBS deutlich verbessert. Abgesehen von leichten Fällen sollten die Patienten auf einer Intensivstation behandelt und die Indikationen zur Intubation und Beatmung frühzeitig gestellt werden.
Autonome Funktionsstörungen erfordern eine engmaschige Überwachung der Herz-Kreislauf-Funktion und die großzügige Anlage eines vorübergehenden Herzschrittmachers. Zur Prophylaxe thrombembolischer Komplikationen sind frühzeitige Mobilisierung, Anti-Thrombose-Strümpfe und die Gabe von Heparin notwendig.
Immunmodulierende Therapie
Die Immuntherapie hat zum Ziel, die Rückbildung der Symptome zu beschleunigen und damit die Zeitdauer der maschinellen Beatmung und den Aufenthalt auf der Intensivstation abzukürzen. Während Glukokortikosteroide bei verschiedenartigen anderen neuroimmunologischen Erkrankungen wie z. B. der Myasthenia gravis oder auch der chronischen Polyneuritis wirksam sind, konnte eine große randomisierte, plazebo-kontrollierte Studie bei 242 GBS-Patienten keinen Effekt für eine Pulstherapie mit 500 mg Methylprednisolon über 5 Tage zeigen. Eine systematische Zusammenfassung („Cochrane review“) von sechs randomisierten Studien bestätigte diese Befunde, wobei nach 4 Wochen kein Unterschied zwischen den Behandlungsgruppen festzustellen war, während nach einem Jahr die mit Steroiden behandelten Patienten sogar leicht schlechter abschnitten.
Demgegenüber konnte die Wirksamkeit der Plasmapherese-Therapie in mehreren kontrollierten Studien gesichert werden. Übereinstimmend besserten sich unter einer Behandlung mit 5 Plasmapheresen mit jeweils 50 ml/kg Körpergewicht an alternierenden Tagen die Patienten schneller, konnten frühzeitiger von der künstlichen Beatmung entwöhnt werden und im Durchschnitt einen Monat eher wieder selbständig gehen als die Patienten der Plazebogruppe.
Die Behandlung sollte bei Patienten, die weniger als 5 Meter selbständig gehen können, vorgenommen und innerhalb der ersten zwei Krankheitswochen eingeleitet werden. Kürzlich berichtete eine französische Arbeitsgruppe, dass bei leichter betroffenen Patienten zwei Austauschbehandlungen besser sind als die alleinige unspezifische Therapie, dass vier Behandlungen besser als zwei sind bei Patienten, die nicht ohne Hilfe aufstehen können, und dass bei beatmeten Patienten sechs Plasmapheresen nicht besser als vier sind. Mit den intravenösen Immunglobulinen (IVIg) in einer Dosis von 0,4 g/kg Körpergewicht über 5 Tage steht eine ebenfalls wirksame Therapie als Alternative zur Verfügung. Obwohl eine frühere Studie behauptete, dass IVIg besser als Plasmapherese seien, konnte dieser Unterschied in nachfolgenden Untersuchungen und insbesondere in der bis dahin größten vergleichenden Untersuchung bei 379 Patienten nicht bestätigt werden: Hier waren sowohl der funktionelle Behinderungsgrad nach 4 Wochen als auch verschiedene andere Zielparameter bei allen Patientengruppen gleich, unabhängig davon, ob mit Plasmapherese, IVIg oder einer Kombination beider Therapieformen behandelt wurde.
Damit können beide Therapieverfahren als gleichwertig angesehen werden. Da IVIg technisch einfacher zu applizieren sind, sollten sie bei Patienten ohne Kontraindikationen bevorzugt eingesetzt werden. Inwieweit die zusätzliche Gabe von Glukokortikosteroiden oder ein zweiter Therapiezyklus mit IVIg zu einer zusätzlichen Verbesserung führt, wird derzeit in kontrollierten Studien untersucht.
Selbsthilfegruppen
Die GBS Initiative e.V.
Die Gründung der GBS Initiative erfolgte im Januar 2001. Die Ziele der Initiative sind:
- Unterstützung, Beratung und nachhaltige Betreuung von GBS Betroffenen und deren Angehörige
- Erstellen von geeignetem Informationsmaterial zur Aufklärung über das Guillain-Barré Syndrom und Varianten. Hier geht es darum, das seltene Guillain-Barré Syndrom bekannter zu machen.
- Flächendeckendes Einrichten und Organisieren von örtlichen Gesprächskreisen im deutschsprachigen Raum von Europa und Polen.
- Die GBS Initiative e.V. fördert ausdrücklich die Gründung von Landes- und Ortsverbänden im Sinne einer GBS Selbsthilfegruppe.
- Unterstützung Erfolgversprechender Forschungsprojekte, die GBS betreffen. Im Wesentlichen soll hier die Ursachenforschung unterstützt werden.
- Einen medizinisch wissenschaftlichen Beirat zur Erfüllung dieser Aufgaben einzusetzen.
- Organisation von Fortbildungsveranstaltungen für Repräsentanten und Patienten.
- Enges Zusammenarbeiten mit internationalen GBS Organisationen zum Zwecke des Erfahrungsaustausches.
- Zusammenarbeit mit nationalen Selbsthilfedachorganisationen, um auf rechtliche Maßnahmen Einfluss zu nehmen.
- Einrichten und Betreiben von WEB orientierten Diskussionsforen für die unterschiedlichen GBS Varianten.
Über die GBS-CIDP-Initiative e.V. (info@gbs-selbsthilfe.de) erhalten Sie Informationen zu den Selbsthilfegruppen.
Kontakt
Deutsche GBS CIDP Selbsthilfe e.V.
Oboensteig 4
13127 Berlin
info@gbs-selbsthilfe.de
Telefon: 01525 4211427